Atividade Remota - Química Geral I
Programação
-
MODULO 1
PROFESSOR RESPONSÁVEL : PAULO NAGIPE
-
MODULO 2

PROFESSOR RESPONSÁVEL; RODRIGO RODRIGUES DE OLIVEIRA
-
ALTERAÇÕES NO CRONOGRAMA DA DISCIPLINA
-
 MODULO 3
MODULO 3PROFESSOR RESPONSÁVEL : MARIA RAQUEL GARCIA VEGA
-
-
-
-
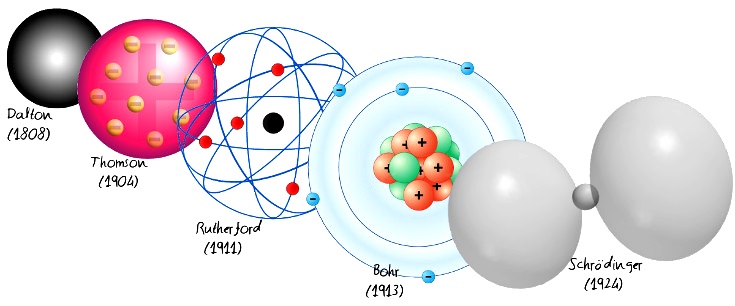
AULA 3 -LIGAÇÕES
Terça-feira, 27 de abril · 8:00 até 10:00pm
Informações de participação do Google Meet
Link da videochamada: https://meet.google.com/gbd-uehm-zjs -
ESTA AVALIAÇÃO TERÁ 3 HORAS DE DURAÇÃO : DAS 21:00 AS 24:00 h.
NÃO SERÃO CONSIDERADOS ARQUIVOS ENTREGUES FORA DO HORÁRIO
SOMENTE SERÃO CONSIDERADOS ARQUIVOS ENVIADOS PARA O EMAIL DA DISCIPLINA: aare.geral.1@gmail.com
-
MODULO 4
PROFESSOR RESPONSÁVEL: JAN SCHRIPSEMA

-
Wait!

NÃO HAVERÁ PROVA NESTA QUINTA-FEIRA EM VIRTUDE DO FERIADO E DA DECRETAÇÃO DO PONTO FACULTATIVO PELO GOVERNO DO ESTADO DO RIO DE JANEIRO. A AVALIAÇÃO OCORRERÁ NO DIA 08/06/2021.
CIBELE
-
MODULO 5
PROFESSOR RESPONSÁVEL : CIBELE STIVANIN

-
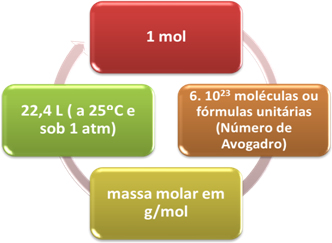
Soluções e cálculos de concentração

-

-
Car@s alun@s,
Em cumprimento ao acordado entre as coordenações do curso, que gentilmente aceitaram a minha proposta, seguem as notas FINAIS do curso após as revisões. Os que se encontram com notas abaixo de seis poderão ter que fazer a prova amanhã. Vamos considerar a norma da graduação desta universidade no que diz respeito a quem pode ou não realizar a avaliação final: quem tiver notas acima de 6,0 está aprovado e está em azul (até o próximo semestre) e não deverá realizar exame final; quem tiver nota final entre 4,0 - 5,9, deverá realizar a prova final (está em vermelho e tachado de amarelo; são apenas 6 alunos); quem tiver nota abaixo de 4,0 não realizará prova final, estando reprovado nesta ARRE.
Qualquer reclamação sobre as notas ou revisões extras, solicitem via requerimento acadêmico às coordenações de cursos, que deverão avaliar, encaminhar aos colegiados acadêmicos e, então para apreciação pela Câmara de Graduação. UMA revisão da avaliação e vista de prova é direito do aluno e dever do professor e, isto foi feito. E, estes pedidos extras podem ser negados em todas as instâncias. Verifiquem se os docentes e coordenações já não estão de férias ou dentro do recesso acadêmico para solicitar isso.
A prova final será realizada no dia 20/07/2021 por questionário na própria plataforma, tal como as demais.
Não custa lembrar, NÃO SOU A RESPONSÁVEL POR NOTAS DE OUTROS PROFESSORES. QUALQUER RECLAMAÇÃO, ENTREM EM CONTATO COM OS COORDENADORES DOS MÓDULOS.
-