Química Geral II - Alunos da Agronomia
Programação
-

Boa tarde a todxs!
No cronograma apresentado aqui vocês irão encontrar as aulas que serão dadas a vocês em cada dia letivo. Também estão marcadas as avaliações, testes e provas, até a prova final.
Desta forma, vocês poderão se programar e estabelecer uma rotina de estudos que permita a compreensão dos conteúdos de forma gradativa.
Tenhamos todxs um ótimo semestre juntos.
Cibele
-
Nesta aula iremos abordar os conceitos de Energia, Trabalho e Calor, bem como as suas inter-relações. Bem vindos à primeira lei da termodinâmica - a lei da conservação da energia.

-
Objetivos da aula:
Apresentar as técnicas para medição do calor envolvido nos processos, bem como definir a função termodinâmica entalpia.
-

Fonte da figura: https://www.manualdaquimica.com/fisico-quimica/energia-livre-gibbs.htm, acessado em 06/09/2022.
-
![Equilíbrio químico [resumos e mapas mentais] - Infinittus](https://infinittusexatas.com.br/wp-content/uploads/2021/05/equilibrio-quimico-mapa-mental-1.jpeg)
Fonte da Figura: https://infinittusexatas.com.br/equilibrio-quimico-resumos-e-mapas-mentais/, acessado em 06/09/2022.
-

Fonte da Figura: https://descomplica.com.br/artigo/ja-ouviu-falar-em-equilibrio-ionico/4Q9/, acessado em 03/10/22.
-
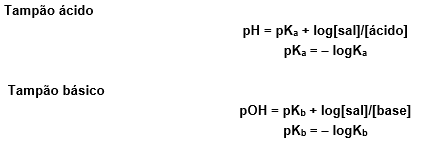
Fonte: https://descomplica.com.br/d/vs/aula/solucao-tampao/ acessado em 03/10/2022
-
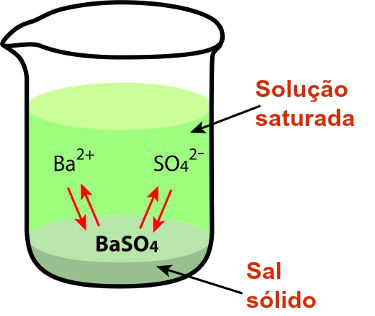
Fonte: https://mundoeducacao.uol.com.br/quimica/constante-produto-solubilidade.htm
-
Alunos (as):
Este é o teste com questões relativas ao conteúdo visto nas últimas aulas, produto de solubilidade.
A avaliação é composta com 8 questões e você só terá uma única chance para resolvê-las. Se passar para a questão seguinte, não há como voltar a anterior. Há apenas uma resposta para cada questão. A avaliação ficará disponível por um período de 24 h para ser realizada, porém ao iniciar, você terá 3 horas corridas para concluir. Tem que iniciar e terminar neste prazo, não há como começar, parar e depois voltar.
O prazo começa no dia 03/11/2022, às 13:00 e termina no dia 04/11/2022, às 12:59. Vejam o melhor horário para realização da avaliação, lembrando que ela não é obrigatória.
Bons estudos,
Cibele

